LAU Study Showcases a Novel AI Model to Accelerate Research in Chemistry
A new AI framework developed by interdisciplinary LAU researchers helps chemists separate truly viable reactions from those that only look promising on paper.

Every medicine and advanced material begins with a carefully orchestrated chain of chemical reactions. Today, artificial intelligence is stepping into this role, proposing “de novo” reactions, transformations generated not by human intuition, but by patterns learned from vast chemical datasets.
The appeal is obvious: A model that can suggest viable transformations that could speed up discovery and expand the options chemists explore. While algorithms can easily generate reactions that look convincing on paper, demonstrating that these reactions are chemically valid—and experimentally feasible—remains far more difficult.
This challenge is the focus of a study titled “Automating Deep Learning-Based Generation and Evaluation of De Novo Chemical Reaction with ChemRxnSAGE” by Computer Engineering graduate Anis Ismail (BE ‘22), supervised by Dr. Joe Tekli, professor at the LAU School of Engineering, in collaboration with Dr. Brigitte Wex, associate professor at the LAU School of Arts and Sciences.
Published in the October 2025 issue of the Journal of Chemical Information and Modeling, the research introduced Chemical Reaction Systematic Assessment of Generation and Evaluation (ChemRxnSAGE), a framework that uses artificial intelligence to generate chemical reactions and then systematically score them for validity and diversity through a repeatable workflow.
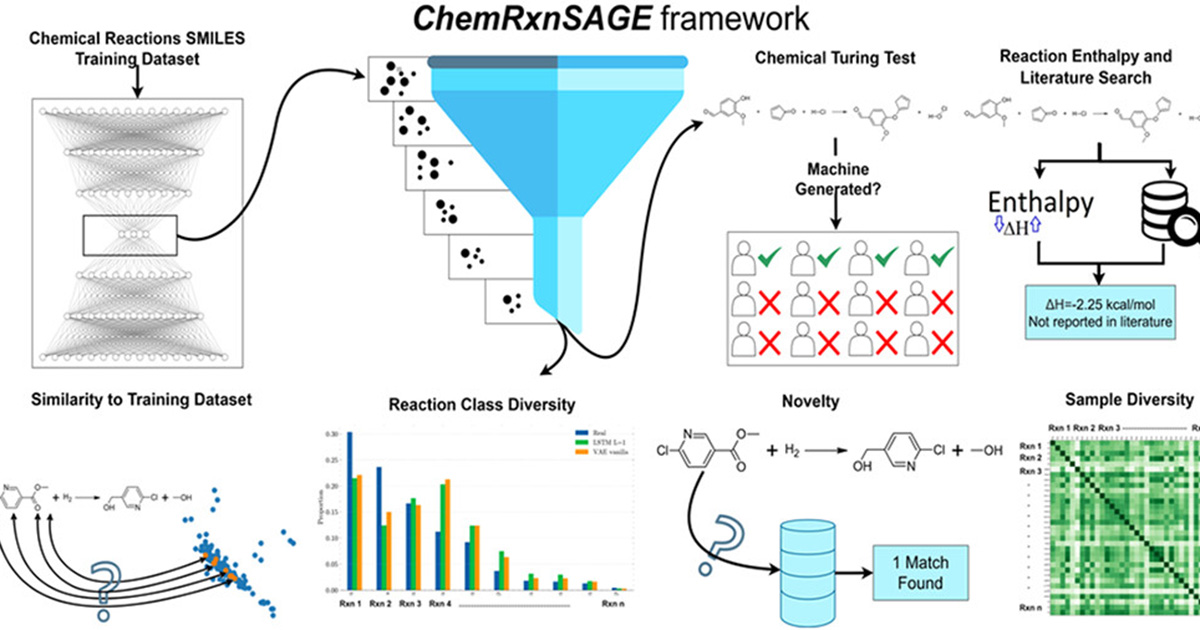
To develop and test the framework, the team applied the pipeline on a data set of 50,000 chemical reactions. Validation combined automated screening with expert judgment in a controlled evaluation, in which chemists assessed a randomized mix of machine-generated and real reactions, allowing the researchers to compare algorithmic output directly with established chemical knowledge.
The automated stage is where ChemRxnSAGE is most distinctive. Because chemistry literature mostly reports successful reactions, clear examples of what does not work are scarce, so filtering becomes essential. The framework rejects malformed reactions, introduces elements that were not present in the reactants, or applies illogical transformations. Across reaction classes, this screening removed about 40 to 70 percent of generated reactions, cutting out many outliers.
“Our main challenges were representing reactions in a model‑friendly yet chemically accurate way, preventing the deep learning model from simply memorizing data, reducing invalid outputs, and balancing novelty with feasibility,” noted Dr. Tekli, which the researchers addressed by developing ChemRxnSAGE.
After filtering, the evaluation asked whether models genuinely explored new chemical space or merely echoed their training data. The results pointed strongly to the former. Nearly 99 percent of the generated reactions were not exact copies of training reactions, and about 99 percent were unique within the generated set. Diversity metrics were also high, suggesting the models did not collapse into repeating the same reaction. Importantly, these automated measures remained stable across five independent runs using different random seeds, underscoring the robustness of the framework.
Human evaluation added a second layer of security. Six experts in organic and synthetic chemistry reviewed a set of 40 reactions. The proposed model produced the highest share judged chemically valid, 46.67 percent, and its reactions were most often mistaken for human-generated, though the reference set of real reactions still scored higher at 68.33 percent valid. For those reactions experts deemed valid, the team ran thermodynamic feasibility checks and found that all shortlisted reactions released energy.
Dr. Wex provided her insights on the properties and behavior of the newly modeled chemical reactions, which allowed the researchers to evaluate the framework. “My role in this project,” she said, “came about since we routinely deploy density functional theory (DFT)—a computational modeling approach—to investigate electronic structures and chemical reactions.”
Commending Ismail’s handling of the bulk of the technical work for the study, Dr. Tekli added that as a result of that and “his impressive university performance as a whole, Anis was admitted to the graduate program of the Catholic University of Leuven, Belgium, where he is currently conducting his PhD in the Laboratory of Multi-omic Integrative Bioinformatics.
Overall, this study positions ChemRxnSAGE as a practical step toward reproducible evaluation in reaction generation. By enabling consistent comparisons across models and highlighting the most promising candidates, the framework helps researchers focus experimental efforts on ideas most worth testing in the laboratory.
To browse more scholarly output by the LAU community, visit our open-access digital archive, the Lebanese American University Repository (LAUR).